Espacios. Espacios. Vol. 30 (2) 2009. Pág. 27
Desiree Moraes Zouain * y Marco Antonio Grecco D’Elia**.
Recibido: - Aprobado
The proposed procedure is shown in Table 6.
Table 6
Procedure to access the CE mark for electrical medical equipments
| STEP | COMPLEMENTARY INFORMATION |
|---|---|
| 1) Verify if the equipment is a Medical Device MD against the definition of Medical Device Directive MDD 93/42 and if it is the use intended by the producer. | Medical Device MD: any instrument, apparatus, appliance, material or other article, used isolated or combined, including the logical supports necessary to its good functioning, intended by the producer to be used on human beings for the purpose of: diagnosis, prevention, monitoring, treatment or alleviation of disease, or injury or handicap; diagnosis, monitoring, treatment, alleviation or compensation of injury or handicap; investigation, replacement or modification of the anatomy or of a physiological process; or control of conception; and which does not achieve its principal intended action in or on the human body by pharmacological, immunological, or metabolic means, although its function can be supported by these means. |
| 2) Verify if there are other European directives applied to the equipment beyond MDD 93/42 and, if positive, identify the harmonized standards to be followed to satisfy the essential requirements. | In the site www.newapproach.org is possible to find the directives and the harmonized standards. The most rigorous directive must be attended to each product, complemented with additional aspects of other applied directives. |
| 3) Classify the equipment according to the annex IX of MDD 93/42 (classes I, IIa, IIb, III). | There are 18 rules on Annex IX of MDD 93/42 which define the classification. |
| 4) Show the attendance of essential requirements (annex I of MDD 93/42) applied to the equipment. | The annex I of MDD 93/42 lists the safety essential requirements to be attended. The annex gives few indications about how the requirements have to be attended. The European harmonized standards are more practical to producers, since they describe testing procedures and limit values. The harmonized standards are voluntary applied, but when the producer use the standard have the benefit of conformity presumption, or, if the MD is in accordance with the standard, do attend the essential requirements, too. In the case of MD, the standards to be followed are: General Standard EN IEC 60601-1: General requirements - Part 1 Safety general requirements; collateral standards EN IEC 60601-1-X and particular standards EN IEC 60601-2-XX to the type tests. |
| 5) Adequate the company quality system management according to standard EN ISO 13.485:2003. | The standard EN ISO 13.485:2003 Medical Devices Quality System Management Regulamentar requirements establish the quality systems specific requirements to medical devices and equipments, based on Standard ISO 9001:2000 Quality system management Requirements. |
| 6) Process and document the analysis and management of potential risks that MD can offer to patients, users and others. | It is recommended to follow the standard EN ISO 14.971:2007 Medical Devices Medical devices risk management appliance, which consists on to foresee the potential risks related to incorrect use, bad functioning, energy transmitted by device, toxicity, contamination, etc.. |
| 7) Adequate the product according to the European harmonized standards requirements. | a) Consult technological institutes and laboratories that have specialized professionals with experience on electrical medical equipment and related technology, who can analyze the devices and to suggest improvements.
b) Realize the necessary tests to verify and assure that every essential requirement is satisfied. There are public founds and financing programs to small and medium companies that supports financially the development and improvement of medical devices. |
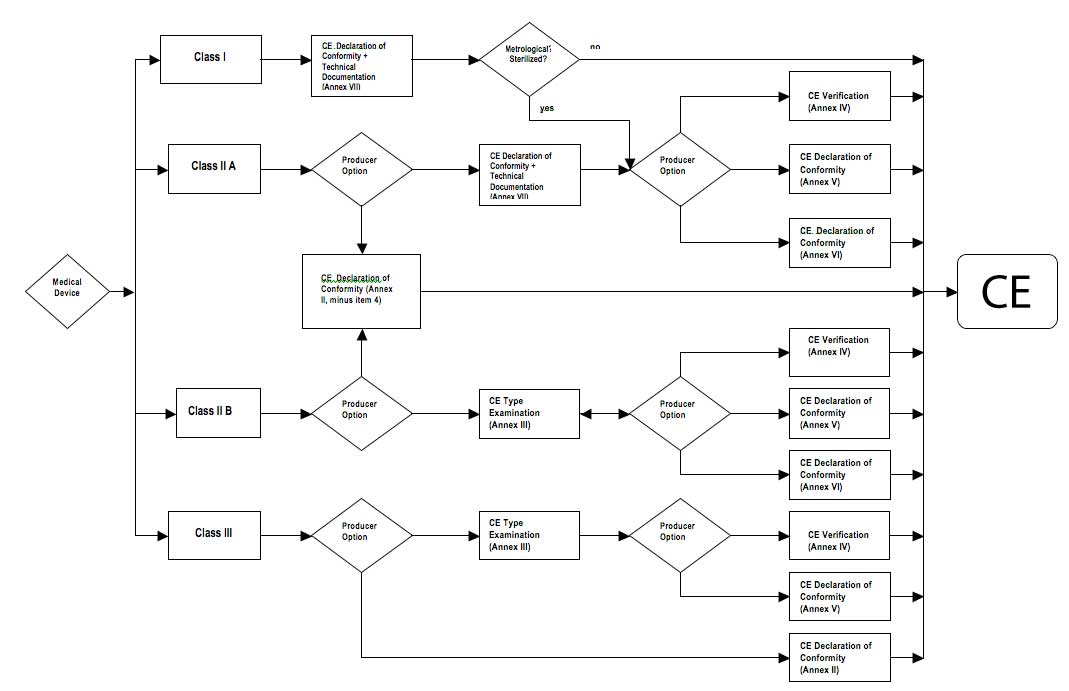
| 8) Choose a scheme, among those allowed according to the classification, to the conformity assessment (Article 11º of MDD 93/42). | This aspect is relatively opened. These procedures are described on the Annex II and VII of MDD 93/42 and are graphically represented on Figure 2. |
| 9) Audit the product and quality system, if applicable. | Contact a Notified Body to perform the the product and quality system audit, if applicable, according to MD classification and the conformity assessment scheme chosen. |
| 10) Prepare the MD technical documents (technical file). | The technical file must contain: general description of equipment; draws, electric schemes, process chart, assembly instructions, etc.; descriptions and orientations necessary to understanding the draws, schemes, charts, diagrams and equipment operation; to sterilizeable products, a description of the used method; relation of applied standards, in part or full, with a description of solutions adopted to satisfy the essential and relevant requirements of directives; master list of draws; life-time of the equipment determined by the producer; the potential risk analyses and management; tests of the equipment and its parts, realized by the producer or in other laboratories; data (clinics, literature, clients researches, etc.) to demonstrate the equipment efficacy and efficiency; packaging and instructions manual; and conformity declaration. |
| 11) Register the legal responsible by the MD in the market. | The producer or its authorized representative, established on the European Union, have to inform the Member-State competent authority, where is localized the site, the address of the site and the description of the equipment focused. |
| 12) Prepare and sign the Conformity Declaration, according to the chosen assessment scheme. | Before the MD is put into the market, the legal responsible prepare and sign a declaration informing the MD is conform all requirements established on applied directives. The declaration has to be prepared according to the chosen conformity assessment scheme. |
| 13) Put CE mark on the equipment. | The marking consist in to put the letters CE on the device, or eventually, if it is not possible because the device size or function, on the packaging or using instructions. Together with the CE have to be put the Notified Body identification number, which is responsible by the procedures foreseen on annexes II, IV, V e VI. |
| 14) Keep a vigilance system to the MD commercialized. | Any incident happened after the MD is put into the market has to be informed to the same competent authority. |
Source: Prepared by the authors
The diagram on Figure 2 shows the electrical medical equipments conformity assessment options that the producers have to choose, according to Article 11 of Medical Devices Directive - DDM 93/42 (CCE, 1993).
Figure 2
Conformity assessment scheme for Medical Devices, according
to Article 11º of MDD 93/42 (the mentioned annexes are from MDD93/42)
Source: Adaptation of Directive 93/42/CEE (1993)
To validate the proposed procedure, it was submitted to four specialists, the same who participated on the survey questionnaire validation. To these specialists was asked to evaluate if the procedure information and orientation are correct, if they are in adequate manner, if they attend to the necessities of the small and medium producers of electrical medical equipments, and if these companies would have benefits with this procedure. Suggestions to improve the procedure were asked too. The specialists considered the proposed procedure a useful tool to the small and medium producers of electrical medical equipments, as an important guide to the process of getting the CE mark.